Research
Key aspects of our research are theoretical studies on transport and relaxation processes in condensed matter, on nucleation and growth phenomena on surfaces, and on basic problems in non-equilibrium statistical mechanics. For our studies we use different analytical and numerical methods in combination with computer simulations.
Growth kinetics on surfaces
Nucleation kinetics in epitaxial growth
We investigate the nucleation and growth of clusters on surfaces that emerge during evaporation of atoms on solid substrates. The focus of our studies is to understand the onset of formation of stable clusters on preexisting islands, which is of crucial importance for the resulting film morphology. The problem is treated by various means, such as kinetic Monte-Carlo simulations, scaling concepts, analytical calculations, and numerical solutions of newly developed rate equations. The theoretical concepts are based on the calculation of lifetimes and rates for the relevant states and their transitions, respectively. Currently we study the growth of alloys under co-deposition of their components, the self-organization and self-assembly of organic molecules on inert dielectric surfaces, and fluctuation methods for the determination of diffusion coefficents and tensors of single molecules on surfaces by scanning probe tips.
Structure and kinetics of polymer blends on surfaces
Simulation of the demixing of a polymer blend on a surface
For many applications it is important to understand polymer dynamics on semi-macroscopic time scales, corresponding to configurational changes of polymer chains on length scales comparable with or larger than the radius of gyration. A problem of active current research in nano-technology, for example, is the tailoring of thin polymer films on surfaces. Spontaneous phase separation processes of incompatible polymer blends may be used to translate a chemical pattern on the surface into a pattern of varying polymer compositions. In order to gain insight into the demixing processes and optimal conditions for pattern translation, we investigate soft ellipsoid models for polymeric systems. In these models the polymers are mapped onto inter-penetrating ellipsoids that can change their shape, position and orientation. The probability for a particular shape follows from an intramolecular free energy functional. A monomer density is assigned to each ellipsoid, and the interaction results from the overlap of the respective densities. An important feature of the ellipsoid model is that the input parameters can be determined from microscopic models appropriate to describe the polymer system on short time scales.
Ion transport in glasses
Sketch of the motion of akali ions in a silicate glass
Traditional glasses are composed of a network former, for example silicate or borate, and a network modifier usually being an alkali oxide. In the glassy phase the alkali ions are decoupled from the frozen silicate or borate host network and have a relatively high mobility. More complex glass compositions have been developed in recent years to obtain specific material properties. These are useful for the design of various technical devices such as batteries, chemical sensors or optical fibers. From the theoretical point of view, the understanding of ion dynamics in glasses is a challenging task due to the existence of various anomalies. Dramatic variations over many orders of magnitude are observed in transport coefficients (tracer diffusion coefficients, dc-conducutivity, etc.), if the concentration of mobile ions is changed, or if one type of mobile ion is successively replaced by another type of mobile ion (mixed alkali effect). The time-dependent ion dynamics, which can be explored by various experimental probes (frequency dependent conductivity, quasielastic neutron scattering, nuclear spin lattice relaxation, internal friction), is characterised by correlation functions that decay slower than exponential ( non-Debye relaxation). Based on our previous work, it is our aim to develop a theory that allows one to explain both the variations in ion mobilities upon changes in the composition of mobile ions and the non-Debye dynamics within a unified description.
Slow dynamics in complex systems
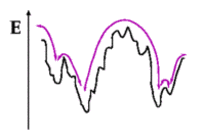
Illustration of effective transitions in a complex energy landscape
Dynamical processes in non-equilibrium systems are not invariant with respect to time translations. Correlation and relaxation functions characterising the dynamics of these systems depend on two times: The observation time of the correlation or relaxation being measured, and the waiting time elapsed since the system was brought into the non-equilibrium state. For observation and waiting times much larger than microscopic time scales (e.g. for molecular vibrations) one finds in many different complex systems (glasses, polymers, colloidal aggregates, pinned interfaces, vortex lines in superconductors) that the two-time correlation and relaxation functions can be described by a function of one variable only, which results from a particular combination of the observation and waiting time. To explain generic features of these physical aging processes, we perform computer simulations and analytical calculations of simple models that capture the essential features of the phenomena. Currently, we are interested in the question if and how a generalized fluctuation-dissipation theorem can be formulated in the non-equilibrium state. A further aim is to describe rejuvenation and memory effects observed in temperature and field cycling experiments.
Density functional theory for lattice systems
To describe surface-phase and wetting transitions as well as confinement effects on bulk phase transformations, density functional theory has proven most successful. Discrete (lattice) versions of density functional theory in particular can be applied to metallic alloys, adsorbed submonolayer films, intercalation compounds, crystalline ionic conductors etc. By using an approach based on Markov chains, we develop methods to derive exact functionals in one dimension that serve as a guideline for constructing approximate functionals in higher dimensions. Density functionals for fluid systems can be obtained by taking appropriate continuum limits. By employing time-dependent density functional theory, kinetic equations for ordering phenomena are derived that compare much better with Monte-Carlo simulations than ordinary mean-field equations. Phenomenolgical Onsager coefficients entering standard kinetic equations can be expressed in terms of microscopic local correlation functions.
Statistical analysis of physiological signals
Novel ideas to simplify and improve the diagnosis of deseases are based on the analysis of time correlations and space-time patterns in physiological signals with methods borrowed from Statistical Physics. In this new project we currently focus on two problems. In the first problem we try to develop automatic computer-based procedures to identify the anaerobic threshold in ergo-spirometer data sets. In the second problem, we develop soft- and hardware tools to extract various physiologic data sets (ECG, blood pressure, oxygen saturation, etc.) continously from monitoring systems used at intensive care sections of hospitals. The aim of this work is to test recently proposed anlaysing methods with respect to their potential power in forecasting sudden functional breakdowns in the human body.
